Home | Projects | CPGE | My carbon footprint
Introduction
Electronic correlation is the interaction between electrons in the electronic structure of a quantum system. The correlation energy is a measure of how much the movement of one electron is influenced by the presence of all other electrons.
Density Functional Theory
- First theorem : the ground-state electron density
uniquely determines the external potential acting on the electrons. This means that if the electron density of a system is known, the external potential can be uniquely inferred, allowing for the derivation of all ground-state properties, including the total energy, from the electron density. - Second theorem : for any trial electron density
, the energy functional will yield a value that is greater than or equal to the ground-state energy . The equality holds when the trial density corresponds to the true ground-state density. This variational principle implies that one can minimise the energy functional with respect to the electron density to find the ground state of a system.
Downsides
The Hohenberg-Kohn theorems are limited to ground-state properties, providing no direct insight into excited states, which poses challenges in studying electronic excitations and charge transfer processes.
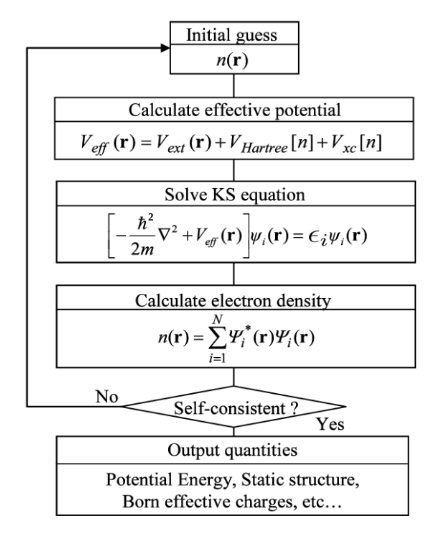
Algorithm
Electron Exchange Correlation
The DFT exchange-correlation potential,
Common approximations
Local Density Approximation (LDA)
is considered to depend solely on the local electron density. This approximation is based on the idea that in regions where the electron density is nearly uniform, the behaviour of electrons can be likened to that of a homogeneous electron gas. - While simple, LDA can perform reasonably well for certain materials, especially solids with nearly uniform electron densities.
Generalised Gradient Approximation (GGA) - Introduces a dependence on the spatial gradients of the electron density. By including information about how the density changes in space, GGA provides more accurate results for systems where the density varies significantly, such as in molecules or surfaces.
In practice
The calculation of
(SCF) method. The DFT algorithm begins with an initial guess for the electron density, usually based on atomic configurations.
- Using this (intial) density, the Kohn-Sham equations are solved to update the potential and electron density
- The exchange-correlation potential,
, is recalculated at each step based on the updated density
This process continues until the electron density converges to a self-consistent solution.
Source : An Introduction to Density Functional Theory, Dr. Mohammad Abdur Rashid